
 Open Access
Open Access Abstract
This paper presented the electronic transport properties of one dimensionally sawtooth – sawtooth edge pentagonal silicon dicarbide nanoribbons (SS-pSiC2) doped n-type (nitrogen: N) atoms and p-type (bore: B) atoms based on the combination of the density functional theory and the nonequilibrium Green’s function formalism. Electronic properties such as the energy band structure (BS), the density of state (DOS) and partial density of state (PDOS) were investigated and compared with the undoped one. The results showed that N played a role in contributing to the Fermi-level optical states that made the system transition from semiconductor to metal. The contribution of the state of the H atom was recognized but this one was very small. When doping B, the material system possessed the metallic properties because the Fermi level crossed the valence subregion. With the Fermi level falling to the top of the valence band, the contribution of the valence electronic states of B was mainly near the Fermi leve. This showed that B was a p-type impurity. The electron transport properties were also investigated, specifically the Volt-Ampere characteristic (I‒V curve) of bipolar devices based on SS-pSiC2 which was passivated by H atoms on the boundary. When doping N, the current appeared as soon as the bias voltage had just a small value. This was the Volt-Ampere characteristic of the metal. The current value also tended to saturate when the voltage was high. The current was about two orders of magnitude larger than the undoped case but lower than the current value of SS-PGNR. When doping B, the current value tended to saturate earlier than the case of N doping when the voltage was high. The current was about an order of magnitude smaller than the n-type doping case. This meant that the amperage obtained with a predominantly hole carrier was smaller than that of a predominantly electron carrier. The results showed that the electronic structure and the current of the studied samples strongly depended on the doping element as well as on the doping type. When doping, the studied system changed from the semiconductor to metal. Furthermore, with p-type doping, the flow of mainly hole carriers was one order of magnitude lower in intensity than that of mostly electron carriers (n-type doping).
MỞ ĐẦU
Năm 2004, Andre Geim và Konstantin Novoselov đã tách thành công một lớp đơn nguyên tử carbon từ khối graphite bằng phương pháp bóc tách cơ học 1 . Điều này đã chứng minh sự tồn tại của một loại vật liệu hai chiều (2D) được cấu thành từ carbon và được gọi là graphene. Với graphene, các nguyên tử carbon sắp xếp theo mạng hình lục giác. Graphene ở trạng thái không gắn trên đế, các hạt tải của nó có độ linh động vào khoảng 230.000 cm 2 /V.s ở nhiệt độ thấp và 120.000 cm 2 /V.s ở 240 K 2 . Bên cạnh những tính chất điện tử đặc biệt là độ linh động cao, dẫn điện tốt thì graphene còn có những tính chất nổi trội khác như độ bền cơ học cao, là vật liệu siêu nhẹ và dẫn nhiệt tốt 2 , 3 . Với những tính chất vật lý ưu việt và vượt trội của nó, các vật liệu hai chiều đã gây ra sự hấp dẫn cho nhiều nhà nghiên cứu lĩnh vực vật liệu.
Gần đây, enta-Graphene (PG), một cấu trúc các nguyên tử C đơn lớp xếp hình ngũ giác đã được đề xuất. Cấu trúc PG là tổ hợp của các nguyên tử C1 (lai hóa sp 3 ) và các nguyên tử C2 (lai hóa sp 2 ) 4 . Các nghiên cứu tiên đoán PG là một bán dẫn có vùng cấm gián tiếp với độ rộng vùng cấm là 3,25 eV 5 , 6 . PG có tính bán dẫn nội tại. Bên cạnh đó, các nghiên cứu này cũng tiên đoán rằng PG có hệ số Poisson âm bất thường, độ bền siêu cao và khả năng chịu được nhiệt cao tới 1.000 K, vượt trội so với graphene. Vào năm 2016, Xia và cộng sự 7 sử dụng phương pháp lắng động hóa pha hơi đã xác định rằng họ có thể nuôi định hướng lớp PG lớn trên lá Cu. Tiếp theo sau nhiều vật liệu cấu trúc đơn lớp dạng ngũ giác đã được đề xuất, thí dụ như pentagonal silicon dicarbide (p‒SiC 2 ) and pentagonal carbon nitride (p‒CN 2 ) 8 .
Nhằm mục đích giảm kích thước linh kiện, tấm PG được cắt thành các cấu trúc giả một chiều được gọi là enta-graphene nanoribbon (PGNR). PGNR có 4 loại: ZZ, AA, ZA, SS. Để ổn định cấu trúc, tắt dần các liên kết dư, các mẫu được thụ động biên bởi nguyên tử H. Trong đó, SS là loại đáng chú ý nhất không chỉ bởi vì nó là bán dẫn mà còn vì nó là cấu trúc bền nhất trong bốn loại PGNR. Sự hấp phụ các phân tử khí CO, CO 2 , NH 3 trên bề mặt cấu trúc này đã được nghiên cứu, xác định được đặc tính hấp phụ của chúng 9 . Nghiên cứu tính chất vận chuyển điện tử của SSPGNR pha tạp thay thế (Si, P, N) và điều chỉnh biên cấu trúc đã được thực hiện, kết quả cho thấy cấu trúc vùng năng lượng và đặc trưng I‒V của SSPGNR pha tạp P và N thay đổi đáng kể so với mẫu nguyên sơ 10 , 11 . Một hiện tượng cũng rất đáng chú ý là cấu trúc PGNR có tính đối xứng thấp hơn graphene nên việc pha tạp vào các vị trí khác nhau trong vòng năm nguyên tử carbon cũng thu được các đặc tính điện tử và vận chuyển điện tử khác nhau 12 .
Với những đặc tính thú vị ấy của cấu trúc ngũ giác, bằng việc tính toán dựa trên lý thuyết phiếm hàm mật độ, sự ổn định cấu trúc và đặc tính điện tử của các dãy nano p-SiC 2 dạng ngũ giác (p-SiC 2 NRs) đã được nghiên cứu một cách có hệ thống. Kết quả nghiên cứu cho thấy p-SiC 2 NRs ổn định cấu trúc nhưng bị vênh. Trong bốn dạng cấu trúc p-SiC 2 NRs (ZZ, ZA, AA và SS) thì cấu trúc SS-p-SiC 2 NR được xác định ổn định nhất về mặt nhiệt học và động lực học và có tính bán dẫn 13 . Tuy nhiên, tính chất điện tử và vận chuyển điện tử của cấu trúc SS-p-SiC 2 NR pha tạp chưa được nghiên cứu, vì thế, bài báo này trình bày việc nghiên cứu một cách có hệ thống tính chất điện tử và vận chuyển điện tử của SS-p-SiC 2 NR pha tạp hai nguyên tử tạp đại diện loại n (N) và loại p (B) trên cơ sở lý thuyết phiếm hàm mật độ kết hợp với phương pháp hàm Green không cân bằng. Các kết quả nghiên cứu góp phần định hướng thực nghiệm và khả năng ứng dụng của hệ vật liệu cho linh kiện vi điện tử 14 , 15 .
VẬT LIỆU VÀ PHƯƠNG PHÁP
Các tính chất điện tử và vận chuyển điện tử của SS-pSiC 2 pha tạp nguyên tử tạp loại n (nitrogen-N) và loại p (boron-B) thể hiện trên Figure 1 , được nghiên cứu bằng cách sử dụng lý thuyết phiếm hàm mật độ kết hợp với hình thức luận hàm Green không cân bằng 16 , 17 .
ự đa dạng của các thuộc tính điện tử và sự vận chuyển điện tử của SS-pSiC 2 một chiều bằng cách pha tạp được lưu ý qua việc nghiên cứu các đặc tính điện tử và vận chuyển của SS-pSiC 2 thụ động biên H, và pha lần lượt hai nguyên tử tạp khác nhau vào vị trí gần biên. Tất cả các mẫu được nghiên cứu đều được tối ưu hóa bằng cách sử dụng phép tính DFT trong phép gần đúng GGA của Perdew-Burke-Ernzerhof (PBE) 18 , 19 . Hơn nữa, 300 K được đặt cho nhiệt độ Fermi và lưới k-point Monkhorst–Pack 1 × 1 × 21 được sử dụng. Các hàm sóng điện tử được chọn bộ cơ sở zeta kép. Để bỏ qua sự tương tác giữa các ảnh, không gian chân không được đặt với kích thước 15 Å. Quá trình tối ưu hóa hoàn tất khi lực dư trên mỗi nguyên tử nhỏ hơn 0,001 eV/Å.
Figure 1 . Cấu trúc tinh thể SS-pSiC 2 -4 pha tạp nguyên tử tạp là N (hoặc B) với nguyên tử tạp ở biên (màu đỏ) tại vị trí lai hóa sp 3

Các tính chất điện tử như cấu trúc vùng năng lượng (BS), mật độ trạng thái (DOS), và mật độ trạng thái riêng (PDOS) được khảo sát cùng với các đặc tính vận chuyển điện tử, cụ thể là đặc trưng Volt-Ampe (đường cong I-V) của linh kiện hai cực dựa trên SS-pSiC 2 thụ động biên H 20 . SS-pSiC 2 (SS-pSiC 2 -4) được chọn với độ rộng bốn chuỗi nguyên tử để khảo sát, cấu trúc có độ rộng nhỏ thể hiện rõ nét cấu trúc một chiều. Các cấu trúc được tối ưu và tính chất điện tử, vận chuyển điện tử được tính bởi gói ATK 21 .
KẾT QUẢ VÀ THẢO LUẬN
Pha tạp nguyên tử nitrogen
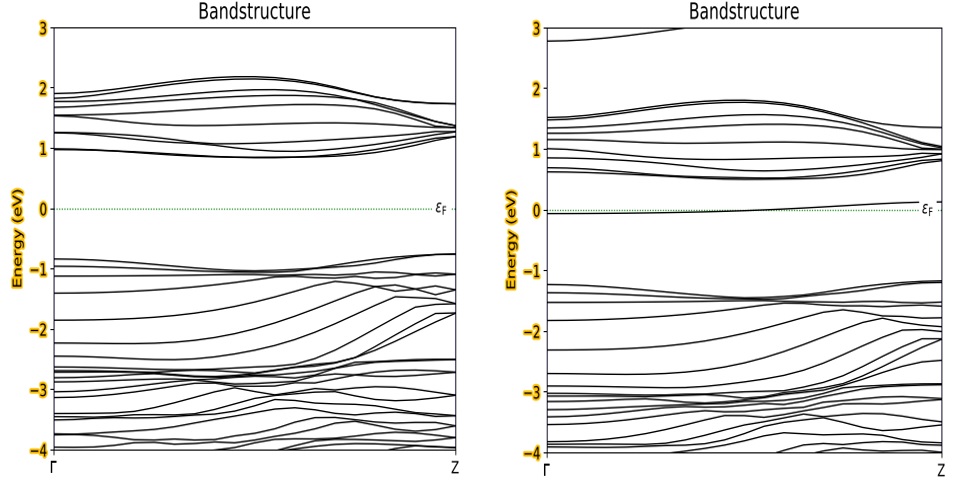
Trong Figure 2 , cấu trúc vùng năng lượng của SS-pSiC 2 pha tạp nguyên tử tạp loại n (nitrogen-N) được trình bày. Trong đó, đường không liền nét màu xanh vẽ mức Fermi. Rõ ràng rằng SS-pSiC 2 -4 thuần có đặc tính bán dẫn, độ rộng vùng cấm vào cỡ 1,9 eV dạng vùng cấm gián tiếp 13 nhưng SS-pSiC 2 pha tạp N sở hữu đặc tính kim loại, mức Fermi cắt ngang vùng dẫn con.
Figure 2 . Cấu trúc vùng điện tử của SS-pSiC 2 -4 chưa pha tạp (hình trái) và pha tạp loại n với nguyên tử tạp là N (hình phải).
Vai trò của tạp N, (PDOS) DOS và DOS được thể hiện trên Figure 3 .
Figure 3 . Mật độ trạng thái DOS (hình bên trái) và mật độ trạng thái riêng PDOS (hình bên phải) của SS-pSiC 2 -10 khi pha tạp loại n với nguyên tử tạp là N. Đường không liền nét màu xanh biểu diễn mức Fermi.
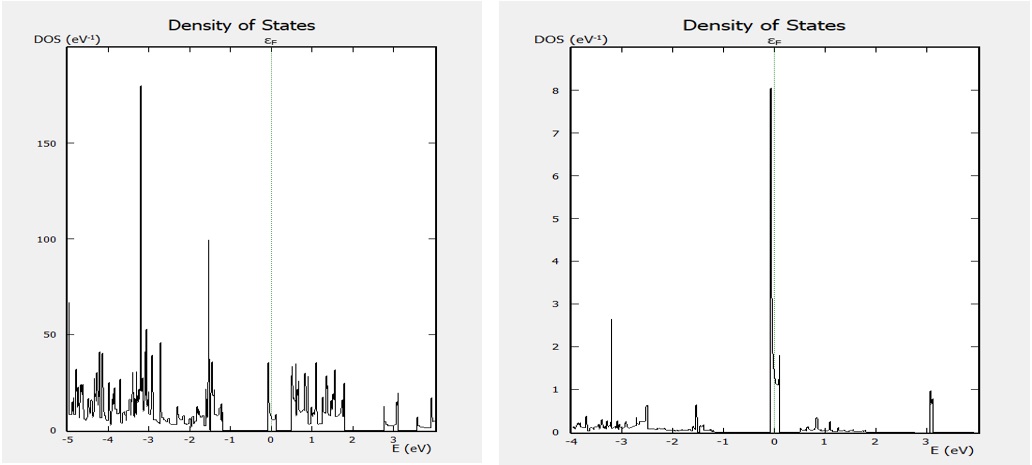
Rõ ràng rằng N có vai trò đóng góp các trạng thái xung quang mức Fermi làm cho hệ chuyển từ bán dẫn sang kim loại với tạp loại n vì mức Fermi dịch chuyển lên vùng dẫn 21 . Sự đóng góp trạng thái của nguyên tử H cũng được ghi nhận nhưng sự đóng góp này là rất nhỏ.
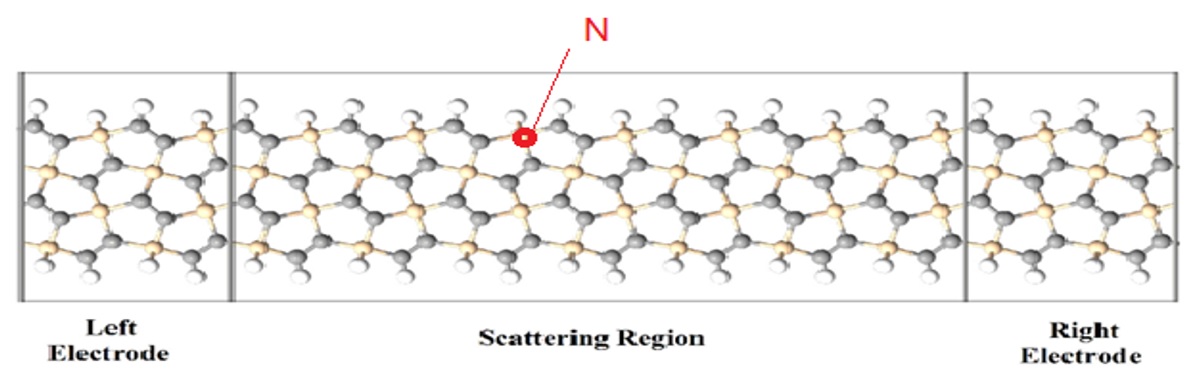
Để khảo sát tính chất vận chuyển điện tử của cấu trúc, linh kiện được thiết kế ( Figure 4 ). Cấu hình tạp đã được tối ưu trước khi gắn vào linh kiện. Đây là linh kiện hai cực với vùng tán xạ trung tâm.
Figure 4 . Minh họa cấu trúc linh kiện hai cực trên nền vật liệu SS-pSiC 2 -4 pha tạp nguyên tử N tại vị trí biên
Các kết quả tính toán về đường đặc tuyến Volt-Ampere của linh kiện được trình bày trên Figure 5 .
Figure 5 . Kết quả tính toán về đường đặc tuyến Volt-Ampere của linh kiện nền vật liệu SS-pSiC 2 -4 pha tạp nguyên tử N tại vị trí biên
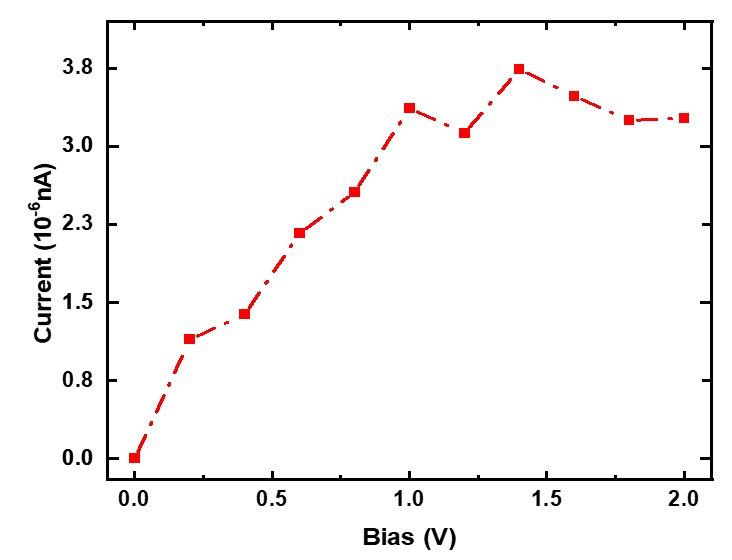
Đối với tính toán này điện áp phân cực được tăng với bước tăng là 0,2 V, để làm thay đổi mức Fermi giữa hai điện cực trái và phải, tạo thế cho vận chuyển điện tử. Kết quả cho thấy dòng điện xuất hiện ngay khi điện áp phân cực có giá trị nhỏ, đây là đặc trưng Volt-Ampere của kim loại. Giá trị dòng cũng có xu thế bão hòa khi điện áp cao. Cường độ dòng lớn hơn trường hợp không pha tạp khoảng hai bậc độ lớn nhưng thấp hơn giá trị dòng của SS-PGNR 10 .
Pha tạp nguyên tử Bo
Tương tự, cấu trúc vùng năng lượng của SS-pSiC 2 pha tạp nguyên tử tạp loại p (boron: B) được trình bày trê Figure 6 . Trong đó, đường không liền nét màu xanh vẽ mức Fermi. SS-pSiC2 pha tạp B sở hữu đặc tính kim loại, vì mức Fermi cắt ngang vùng con hóa trị. Với mức Fermi hạ xuống đỉnh vùng hóa trị thể hiện tính chất tạp loại p.
Figure 6 . Cấu trúc vùng điện tử của SS-pSiC 2 -4 chưa pha tạp (hình bên trái) và pha tạp loại p với nguyên tử tạp là B (hình bên phải).

Vai trò của các điện tử tạp đóng góp vào các trạng thái điện tử quanh mức Fermi bởi vẽ PDOS thể hiện sự đóng góp của tạp B được đánh giá ( Figure 7 ). ự đóng góp các trạng thái điện tử hóa trị của B là chủ yếu gần mức Fermi, thể hiện B là tạp loại p.
Figure 7 . Mật độ trạng thái DOS (hình bên trái) và mật độ trạng thái riêng PDOS (hình bên phải) của SS-pSiC 2 -4 khi pha tạp loại p với nguyên tử tạp là B. Đường không liền nét màu xanh biểu diễn mức Fermi.

Khảo sát tính chất vận chuyển điện tử của cấu trúc nghiên cứu với thiết kế linh kiện như sơ đồ ( Figure 8 ). Cấu hình tạp cũng đã được tối ưu trước khi gắn vào linh kiện. Đây là linh kiện hai cực với vùng tán xạ trung tâm có vai trò của tán xạ tạp.
Figure 8 . Minh họa cấu trúc linh kiện hai cực trên nền vật liệu SS-pSiC 2 -4 pha tạp nguyên tử B tại vị trí biên

Các kết quả tính toán đường đặc tuyến Volt-Ampere của linh kiện được trình bày trên Figure 9 . Đối với tính toán này điện áp phân cực cũng được tăng với bước tăng là 0,2 V. Kết quả cho thấy dòng điện cũng xuất hiện ngay khi điện áp phân cực có giá trị nhỏ, đây là đặc trưng Volt-Ampere của kim loại. Giá trị dòng có xu thế bão hòa sớm hơn trường hợp pha tạp N khi điện áp cao. Cường độ dòng nhỏ hơn trường hợp pha tạp loại n khoảng một bậc độ lớn. Điều này có nghĩa là cường độ dòng thu được với hạt tải chủ yếu là lỗ trống có giá trị nhỏ hơn trường hợp hạt tải chủ yếu là điện tử. Điều này liên quan đến đặc tính của khối lượng hiệu dụng hạt tải.
Figure 9 . Đặc trưng I-V của linh kiện trên nền vật liệu SS-pSiC 2 -4 pha tạp nguyên tử B tại vị trí biên

Các kết quả tính toán thể hiện vai trò của tạp trong đặc tính điện tử và vận chuyển điện tử. Tuy nhiên, giá trị dòng có vẽ nhỏ hơn cấu trúc PG cùng loại. Điều này có thể giải thích rằng, với pSiC 2 là cấu trúc hai loại nguyên tử trong khí đó PG chỉ có một loại nguyên tử C nên tính đồng nhất cao hơn, tán xạ điện tử ít hơn 22 .
KẾT LUẬN
Bài báo trình bày việc khảo sát tính chất điện tử và vận chuyển điện tử của hệ vật liệu giả một chiều SS-pSiC 2 khi được pha tạp các nguyên tử loại n (N) và loại p (B). Kết quả cho thấy cấu trúc điện tử và dòng điện của các mẫu nghiên cứu phụ thuộc mạnh vào nguyên tố tạp cũng như loại tạp pha. Khi pha tạp, hệ nghiên cứu đã chuyển từ bán dẫn sang kim loại thể hiện rõ ở dạng của cấu trúc vùng điện tử và dạng của đặc tuyến I‒V. Kết quả nghiên cứu góp phần hiểu hơn tính chất điện tử của SS‒pSiC 2 mới phát hiện, định hướng thực nghiệm tổng hợp và ứng dụng vật liệu cấu trúc mới này.
LỜI CẢM ƠN
Nghiên cứu này được tài trợ bởi Trường Đại học Thủ Dầu Một trong khuôn khổ đề tài DT.20.2-048.
DANH MỤC CÁC TỪ VIẾT TẮT
sp 2 : lai hóa một quỹ đạo s và hai quỹ đạo p của cùng lớp vỏ nguyên tử để hình thành ba quỹ đạo tương đương
sp 3 : lai hóa một quỹ đạo s và ba quỹ đạo p của cùng lớp vỏ nguyên tử để hình thành bốn quỹ đạo tương đương
SS-pSiC 2 : Penta SiC 2 hai biên răng cưa
DOS: Mật độ trạng thái
PDOS: Mật độ trạng thái riêng
2D: Hai chiều
PG: Penta-Graphene
p-SiC 2 : penta-SiC 2
p-SiN 2 : penta-SiN 2
PGNR: Dải nano Penta Graphene
ZZ: Hai biên zigzac
AA: Hai biên ghế bành
ZA: Biên zigzac - biên ghế bành
SS: Hai biên răng cưa
SSPGNR: Dải nano Penta Graphene hai biên răng cưa
GGA: Gần đúng gradient tổng quát
PBE: Perdew–Burke–Ernzerhof
BS: Cấu trúc vùng
SS-pSiC 2 -4: pSiC 2 hai biên răng cưa rộng 4 chuỗi nguyên tử
ATK: Bộ công cụ Atomistix
XUNG ĐỘT LỢI ÍCH
Các tác giả đồng ý không có bất kỳ xung đột lợi ich nào liên quan đến các kết quả đã công bô.
ĐÓNG GÓP CỦA CÁC TÁC GIẢ
Võ Văn Ớn: phân tích kết quả, viết báo.
Lê Nhật Thanh: thiết kế cấu trúc hệ vật liệu, chạy mô phỏng.
Ngô Vũ Hảo: thiết kế cấu trúc hệ vật liệu, chạy mô phỏng.
Nguyễn Thành Tiên: phân tích kết quả, viết báo.
References
- Novoselov KS, Geim AK, Morozov SV, Jiang DE, Zhang Y, Dubonos SV et al. Electric field effect in atomically thin carbon films. Science. 2004;306(5696):666-9. . ;:. PubMed Google Scholar
- Novoselov KS, Jiang D, Schedin F, Booth TJ, Khotkevich VV, Morozov SV et al. Two-dimensional atomic crystals. Proc Natl Acad Sci U S A. 2005;102(30):10451-3. . ;:. PubMed Google Scholar
- Bolotin KI, Sikes KJ, Jiang Z, Klima M, Fudenberg G, Hone J et al. Ultrahigh electron mobility in suspended grapheme. Solid State Commun. 2008;146(9-10):351-5. . ;:. Google Scholar
- Zhang S, Zhou J, Wang Q, Chen X, Kawazoe Y, Jena P. Penta-graphene: A new carbon allotrope. Proc Natl Acad Sci U S A. 2015;112(8):2372-7. . ;:. PubMed Google Scholar
- Wang Z, Dong F, Shen B, Zhang RJ, Zheng YX, Chen LY et al. Electronic and optical properties of novel carbon allotropes. Carbon. 2016;101:77-85. . ;:. Google Scholar
- Yu ZG, Zhang YW. A comparative density functional study on electrical properties of layered penta-graphene. J Appl Phys. 2015;118(16):165706. . ;:. Google Scholar
- Xia KL, Artyukhov VI, Sun LF, Zheng JY, Jiao LY, Yakobson BI et al. Growth of large-area aligned pentagonal graphene domains on high-index copper surfaces. Nano Res. 2016;9(7):2182-9. . ;:. Google Scholar
- Berdiyorov GR, Madjet MEA. First-principles study of electronic transport and optical properties of penta-graphene, penta-SiC2 and penta-CN2. RSC Adv. 2016;6(56):50867-73. . ;:. Google Scholar
- Tiên NT, Mi TY. Nghiên cứu hiện tượng hấp phụ phân tử khí trên dãy Nano penta-graphene dạng răng cưa. Can Tho Univ J Sci. 2020;56(2):21-9. . ;:. Google Scholar
- Tien NT, Thao PTB, Phuc VT, Ahuja R. Electronic and transport features of sawtooth penta-graphene nanoribbons via substitutional doping. Phys E Low Dimensional Syst Nanostruct. 2019;114:113572. . ;:. Google Scholar
- Tien NT, Bich Thao PTB, Phuc VT, Ahuja R. Influence of edge termination on the electronic and transport properties of sawtooth penta-graphene nanoribbons. J Phys Chem Solids. 2020;146:109528. . ;:. Google Scholar
- Phuc VT, Thao PTB, Ahuja R, Tien NT. Effect of phosphorus doping positions on electronic transport properties in the sawtooth penta-graphene nanoribbon: first-principles insights. Solid State Commun. 2022;353:114859. . ;:. Google Scholar
- Mi TY, Khanh ND, Ahuja R, Tien NT. Diverse structural and electronic properties of pentagonal SiC2 nanoribbons: A first-principles study. Mater Today Commun. 2021;26:102047. . ;:. Google Scholar
- Glavin NR, Rao R, Varshney V, Bianco E, Apte A, Roy A et al. Emerging applications of elemental 2D materials. Adv Mater. 2020;32(7):e1904302. . ;:. PubMed Google Scholar
- Khan K, Tareen AK, Aslam M, Wang RH, Zhang YP, Mahmood A et al. Recent developments in emerging two-dimensional materials and their applications. J Mater Chem C. 2020;8(2):387-440. . ;:. Google Scholar
- Sholl DS, Steckel JA. Density functional theory: a practical introduction. John Wiley & Sons. ISBN: 978-0-470-37317-0; 2011. . ;:. Google Scholar
- Lee JG. Computational materials science: an introduction. CRC press. ISBN 9781498749732; 2016. . ;:. Google Scholar
- Perdew JP, Burke K, Ernzerhof M. Generalized gradient approximation made simple. Phys Rev Lett. 1996;77(18):3865-8. . ;:. PubMed Google Scholar
- Schlipf M, Gygi F. Optimization algorithm for the generation of ONCV pseudopotentials. Comput Phys Commun. 2015;196:36-44. . ;:. Google Scholar
- Tien NT, Thao PTB, Thuan LVP, Chuong DH. First-principles study of electronic and optical properties of defective sawtooth penta-graphene nanoribbons. Comp Mater Sci. 2022;203:111065. . ;:. Google Scholar
- Smidstrup S, Markussen T, Vancraeyveld P, Wellendorff J, Schneider J, Gunst T et al. QuantumATK: an integrated platform of electronic and atomic-scale modelling tools. J Phys Condens Matter. 2019;32(1):015901. . ;:. PubMed Google Scholar
- Tien NT, Thao PTB, Lin MF. Electronic and transport properties of the sawtooth-sawtooth penta-graphene nanoribbons. CRC Press; 2022. p. 29. . ;:. Google Scholar





