
 Open Access
Open Access Abstract
The phonon calculations of hydrogen when adsorbed on the missing row Pt(110)-(1x2) surface using the Ultrahigh Vacuum (UHV) model were studied. The calculations were based on a combination of Density Functional Theory (DFT) and the approximation of hydrogen vibrations on the surface. The harmonic vibration of H on the Pt surface was used for the calculation in this study. At the hydrogen coverage of 1ML for the model surface (when H completely covering the Pt surface), the interaction formed at the edge of the first layer (short bridge - R) was the most stable. The less stable positions were recorded as the bridge position between two atoms on a horizontal row in the second layer (trough - T), the top position of the second surface layer - fcc (F), the bridge position of the second surface layer - hcp (F'), and the least stable position was the top position of the first layer (T'). When accounting the quantum calculation for the model, the adsorption energy of hydrogen at the short bridge position increased by 90 meV, and the stable adsorption order of hydrogen changed with the most stable positions ware R, T, T', F', F respectively. This demonstratesd the significant influence of quantum effects on the adsorption model.
GIỚI THIỆU
Trong gần 3 thập kỉ trở lại đây, điện hóa bề mặt được xem là môn khoa học có ảnh hưởng đến rất nhiều lĩnh vực khác nhau như vi điện tử, xúc tác, pin nhiên liệu 1 , 2 . Trong đó, hydro – platium được xem như là một trong số các mô hình được nghiên cứu chuyên sâu 3 , 4 , 5 , 6 , 7 , 8 , 9 . Mặc dù bề mặt (111) là bề mặt ổn định nhất đối với mô hình tinh thể kim loại lập phương (FCC), các bề mặt khác cũng cần phải được nghiên cứu, vì trong các ứng dụng thực tế, bề mặt xúc tác phân tán nhiều phần khác nhau thành ma trận. Trong các phần khác nhau của kim loại FCC, mặt (111) và (100) đã được chỉ ra là các mặt phổ biến nhất 10 . Các nghiên cứu trước đây chỉ ra rằng các vị trí hoạt động xúc tác là trên các bề mặt này 11 hoặc trên biên giữa các mặt này 12 . Liên quan đến vấn đề này, hai nhóm tác giả Gudmundsdóttir và Skúlason thực hiện các tính toán lý thuyết để làm rõ sử ảnh hưởng của các mặt (111) và (100) 13 , 14 . Họ dựa trên thực tế là bề mặt Pt(110)-(1x2) bị khuyết dãy và xem nó như một mô hình tuần hoàn đại diện cho biên giữa các mặt (111) 13 , 14 . Sau đó, sử dụng thực nghiệm lẫn lý thuyết để nghiên cứu sâu hơn sự hấp phụ của hydrogen lên bề mặt Pt(110) 4 , 15 , 16 , 17 , 18 .
Andrzej khảo sát sự hấp phụ của hydrogen lên bề mặt kim loại Pt(100), (111), (110) trong môi trường acid HClO 4 và H 2 SO 4 bằng phương pháp quét vòng điện thế (cyclic voltammograms – CV) 5 . Họ đã xác định và chuẩn hóa năng lượng Gibbs của quá trình hấp phụ, các tham số tương tác và sự biến thiên của chúng, cùng trạng thái bao phủ bề mặt dùng để tham chiếu, từ đó có thể suy ra các tương tác hút đẩy xảy ra khi hydrogen hấp phụ lên các bề mặt platium đơn tinh thể. Tuy nhiên, các kết quả của Lasia không cung cấp được các tính chất vật lý về liên kết của hydro cụ thể trên bề mặt Pt. Đó là động lực để chúng tôi tiến hành tìm hiểu về mặt lý thuyết bằng các tính toán lượng tử sử dụng phiếm hàm mật độ cho bề mặt Pt(110)-(1x2) khuyết dãy.
Mục tiêu đầu tiên trong nghiên cứu này là xác định năng lượng hấp phụ H ứng với các vị trí trên bề mặt Pt(110)-(1x2) khuyết dãy. Các tính toán sự hấp phụ đẳng nhiệt của Pt(110)-(1x2) được thực hiện thông qua việc sử dụng phần mềm mô phỏng SIESTA. Với sự kiểm tra kĩ các thông số ban đầu dùng trong tính toán bằng cách thông qua sự hội tụ số lớp, số k-points, các dữ liệu năng lượng thu được từ phương pháp phiếm hàm mật độ DFT này có tính chính xác cao. Sau đó, nhóm cho H dao động trên bề mặt Pt(110)-(1x2), tính toán phonon được thể hiện qua hai giá trị là tần số giãn nén và năng lượng điểm không (ZPE).
PHƯƠNG PHÁP TÍNH
Nghiên cứu này áp dụng cách thức tính toán DFT tương tự như cách đã được áp dụng thành công trong nghiên cứu hệ H/Pt(111) 7 . Chúng tôi sử dụng kết hợp tuyến tính các orbital nguyên tử (LCAO) và lược đồ giả thế trong phần mềm mô phỏng SIESTA (Spanish Initiative for Electronic Simulations with Thousands of Atoms) 19 , 20 để tiến hành các tính toán nguyên lý cấu trúc điện tử ban đầu. Tuy nhiên việc sử dụng LCAO trong SIESTA có sự sai số về sự chồng lặp của bộ cơ sở (basis set superposition error -BSSE) nên trong bài nghiên cứu trước đây cho hệ H/Pt(111) với cùng phương pháp đã chỉ ra sai số tính toán cho năng lượng hấp phụ khoảng 1% 7 . Mô hình và vị trí hấp phụ của hydrogen trên bề mặt Pt(110)-(1x2) dùng để tính toán DFT được biểu thị trong Figure 1 . Trong tính toán DFT, hàm tương quan trao đổi được sử dụng là xấp xỉ gần đúng gradient (GGA) Perdew và cộng sự 21 . Vì GGA-PBE cho kết quả mô tả chính xác về phản ứng tuyến tính của khí điện tử đồng nhất, về đặc tính theo tỉ lệ đồng nhất và thế năng đưa ra phẳng hơn 21 .
Figure 1 . Mô hình bề mặt Pt(110)-(3x2) với (R) là vị trí cầu nối giữa hai nguyên tử trên một hàng dọc ở lớp trên cùng (cầu ngắn), (F) là vị trí fcc, (F') là vị trí hcp, (T) là vị trí cầu nối giữa hai nguyên tử trên một hàng ngang ở lớp thứ hai (rãnh) 22
 22" width="300" height="200">
22" width="300" height="200">
Chúng tôi đã áp dụng bộ tham số tính toán SIESTA cơ bản theo chuẩn phân cực kép zeta (DZP) và lưới cắt là 200 Ry. Đồng thời, hàm Fermi Dirac với nhiệt độ điện tử tại 300 K được sử dụng để tính các tích phân vùng Brillouin. Chúng tôi sử dụng giá trị năng lượng 200 meV cho độ dịch chuyển của nguyên tử Pt, qua đó xác định bán kính cắt ứng với từng mô men động lượng góc. Các tham số tính toán chuẩn trong SIESTA cung cấp độ chuẩn xác hợp lý cho kết quả của Pt dạng trần (chưa hấp phụ H) lẫn khi hấp phụ nguyên tử hydrogen 7 . Chúng tôi cũng chọn giá trị 3.9242Å ứng với ô cơ sở thực nghiệm thu được để xây dựng mô hình platinum 23 .
Việc tính toán H hấp phụ lên bề mặt Pt(110) – (1x2) được thực hiện thông qua 3 bước thiết lập cấu hình. Đầu tiên, các mô hình bề mặt (1x2) với số lớp nguyên tử khác nhau từ 4 đến 6 lớp nguyên tử Pt tính từ bề mặt được xây dựng. Ứng với từng trường hợp số lớp, chỉ số điểm k thiết lập trong tính toán SIESTA tăng dần từ (4x4x1) đến (13x13x1) Monkhorst Pack (MP), hay từ 11 đến 85 điểm k, với mục đích tìm sự hội tụ theo số lớp và điểm k. Thứ hai, sau khi chạy chương trình SIESTA và thu được mô hình ổn định của bề mặt Pt(110)-(1x2) dạng trần, chúng tôi cho một nguyên tử hydro hấp phụ lần lượt lên từng vị trí khác nhau trên bề mặt Pt(110)-(1x2). Các vị trí này bao gồm fcc (F), hcp (F’), cầu ngắn (R), đỉnh (T’) và rãnh (T) ( Figure 1 ). Thứ ba, để tính ZPE, nguyên tử hydro được dịch chuyển khỏi vị trí cân bằng theo trục z và xấp xỉ bằng một dao động điều hòa.
KẾT QUẢ VÀ THẢO LUẬN
Sự hội tụ năng lượng
Với mỗi dạng cấu trúc bề mặt khác nhau của Pt thì vị trí, mức độ hấp phụ bền vững của H trên đó cũng khác nhau. Do đó, nhóm chúng tôi bắt đầu bằng việc tính toán năng lượng hấp phụ bề mặt để kiểm chứng những vị trí hấp phụ có thể của H trên bề mặt khuyết dãy Pt(110)-(1x2).
Table 1 thể hiện năng lượng hấp phụ của hydrogen được tính bởi công thức:
với là năng lượng tổng của hệ nguyên tử hydrogen hấp phụ lên bề mặt Pt(110)-(1x2), là năng lượng của một phân tử hydrogen cô lập. là năng lượng hấp phụ của H trên Pt. tại vị trí cầu ngắn (R) được ghi nhận là thấp nhất trong các trường hợp, có nghĩa là vị trí hấp phụ bền vững nhất. Do đó khi khảo sát tính hội tụ chúng tôi chọn vị trí R để xét và một vị trí nữa là F, để dùng làm đối chiếu. Và bảng 2 thể hiện độ dài liên kết tối ưu của các nguyên tử H với các nguyên tử Pt lớp trên cùng.
Để kiểm chứng tính đúng đắn của các thông số lựa chọn trong tính toán DFT, sự hội tụ năng lượng được tính toán cụ thể trong bài. Khi giá trị năng lượng được hội tụ tại một điểm chứng tỏ các thông số lựa chọn có tính chính xác cao và có thể dùng làm số liệu so sánh với thực nghiệm. Mục đích của việc xét nhiều lớp và nhiều điểm k khác nhau là để tìm giá trị các tham số tính toán mà tại đó năng lượng hội tụ.
Năng lượng mà chúng tôi xét sự hội tụ là năng lượng tương quan khi hydrogen hấp phụ lên vị trí cầu ngắn (R) so với vị trí fcc (F) của bề mặt Pt(110)-(1x2). Kết quả năng lượng hấp thụ tìm được ứng với hai vị trí này được thể hiện trong Bảng 3. Trực quan hóa dữ liệu của Bảng 3 thông qua Figure 2 và Figure 3 . Dựa vào Figure 2 có thể thấy khi số lớp tăng đến 5, giá trị (eV) = E tot. (R) – E tot. (F) hội tụ với các điểm k khác nhau. Điều này chỉ ra rằng tác động của hydrogen chỉ ảnh hưởng đến 4 lớp tính từ bề mặt Pt(110) khuyết dãy.
Còn từ Figure 3 cho thấy khuynh hướng hội tụ đang tiến đến khi số điểm k tăng đến (13x13x1) MP.

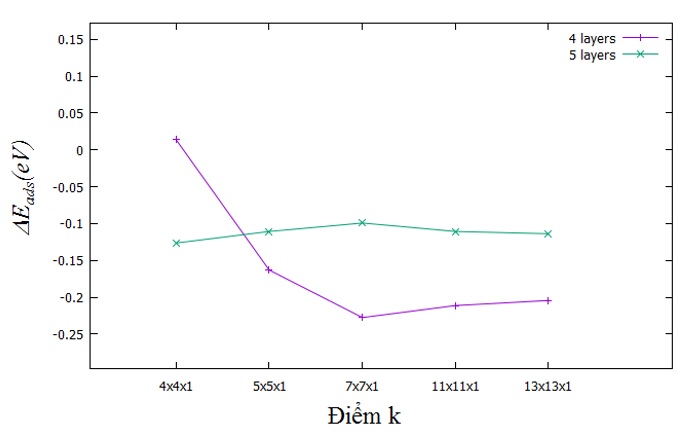
Năng lượng điểm không (ZPE)
Dao động của nguyên tử H xung quanh vị trí cân bằng trên mỗi trục có thể được xem gần đúng là dao động điều hòa vì khoảng cách giữa các nguyên tử Pt và H khá lớn so với dao động của nguyên tử H xung quanh vị trí cân bằng. Do đó, xấp xỉ dao động điều hòa được sử dụng để tính ZPE. Đầu tiên, lấy vị trí hấp phụ sau khi đã ổn định của hydrogen trên bề mặt khuyết dãy Pt(110) – (1x2) là vị trí cân bằng, nhóm tiến hành dao động dịch chuyển nguyên tử hydrogen dọc theo trục liên kết Pt-H một khoảng từ – 0.1 Å đến + 0.1 Å và tính năng lượng của hệ với cấu hình đó. Xấp xỉ năng lượng thu được bằng năng lượng của một dao động điều hòa cho bởi công thức :
với x (Å) là độ dịch chuyển khỏi vị trí cân bằng của nguyên tử hydrogen, hệ số k (eV/Å 2 ) là hằng số lực. Sử dụng phương pháp tính bình phương cực tiểu, chúng tôi suy ra giá trị hằng số lực k và sử dụng nó để tiếp tục tính tần số giãn nén trong tương tác của hydrogen với bề mặt kim loại Pt(110)-(1x2). Biểu thức của tần số giãn nén có dạng như sau:
Trong đó c là vận tốc ánh sáng, m 1 là khối lượng nguyên tử hydro, m 2 là khối lương nguyên tử platium. Cuối cùng, năng lượng ZPE được tính thông qua:
với h là hằng số Planck.
Bảng 4 thể hiện tính toán phonon về hằng số lực, tần số giãn nén cho độ bao phủ H trên Pt là 1ML với số điểm k là (7x7x1) MP và số lớp Pt là 5 lớp. Tần số giãn nén của H trên Pt(110)-(1x2) có giá trị từ 1031 cm -1 đến 2210 cm -1 , giá trị này tương đương với độ giãn nén của H trên bề mặt Pt(111) từ 1184 cm -1 đến 2192 cm -1 7 . Năng lượng hấp phụ ở Bảng 4 được tính bằng việc cộng thêm năng lượng điểm không ZPE vào năng lượng hấp phụ ban đầu E ads từ Table 1 . Như vậy khi tính thêm ảnh hượng lượng tử vào sự hấp phụ của H trên bề mặt Pt(110)-(1x2) thì năng lượng hấp phụ của hệ tăng thêm giá trị từ 64 meV đến 137 meV cho các vị trí hấp phụ khác nhau. Table 1 cho thấy vị trí hấp phụ H bền vững nhất của 5 lớp là R, và kém bền vững nhất là T’, nhưng sau khi tính thêm năng lượng ZPE thì vị trí kém bền nhất lại là F. Điều này chứng tỏ ảnh hưởng lượng tử lên mô hình hấp phụ H/Pt(110)-(1x2) là đáng kể. Trong tương lai, chúng tôi sẽ khảo sát bề mặt Pt lớn để tính toán tương tác liên kết giữa H – H trên bề mặt Pt(110)-(1x2), từ đó cung cấp toàn bộ thông tin về quá trình hấp phụ và ảnh hưởng hấp phụ của hệ H/Pt(110)-(1x2).
KẾT LUẬN
Sử dụng lý thuyết phiếm hàm mật độ DFT và xấp xỉ GGA, phonon của sự hấp phụ hydrogen lên bề mặt khuyết dãy Pt(110)-(1x2) đã được nghiên cứu. Các tính toán hấp phụ cho thấy H bền vững nhất ở vị trí cầu ngắn R. Năng lượng hấp phụ của hydrogen ở vị trí R tăng thêm ~ 90 meV khi tính thêm ảnh hưởng lượng tử lên mô hình. Xa hơn, mô hình tính toán sẽ được mở rộng và khảo sát các tương tác H – H trên bề mặt Pt(110)-(1x2).
LỜI CẢM ƠN
Nghiên cứu này được tài trợ bởi Quỹ Phát triển khoa học và công nghệ Quốc gia (NAFOSTED) trong đề tài mã số 103.01-2017.50.
DANH MỤC TỪ VIẾT TẮT
UHV: Ultrahigh Vacuum: siêu chân không
DFT: Density Functional Theory: lý thuyết phiếm hàm mật độ.
ZPE: Zero Point Energy: năng lượng điểm không.
ML: Mono Layer: đơn lớp.
SIESTA: Spanish Initiative for Electronic Simulations with Thousands of Atoms: gói tính toán mô phỏng.
GGA: xấp xỉ gần đúng gradient.
PBE: Perdew, Burke và Ernzerhof.
MP: Monkhorst Pack.
XUNG ĐỘT LỢI ÍCH
Tác giả không có bất kì xung đột lợi ích nào
ĐÓNG GÓP CỦA CÁC TÁC GIẢ
Nguyễn Văn Hoá: viết bản thảo bài báo, tính toán năng lượng điểm không, năng lượng dao động của hệ H/Pt (110)-(1x2).
Nguyễn Minh Phi: Trực quan hoá mô hình, và tính hội tụ điểm k.
Nguyễn Ngọc Thiện: tính toán sự hấp phụ của H lên các vị trí bề mặt.
Châu Minh Đạo: xây dựng mô hình mô phỏng Pt.
Trần Thị Thu Hạnh: hướng dẫn, định hướng cho toàn bộ nghiên cứu. Kiểm chứng lại các kết quả tính toán, chỉnh sửa và nộp bản thảo.
References
- Markovic NM, Grgur BN, Ross PN, Temperature-dependent hydrogen electrochemistry on platinum low-index single-crystal surfaces in acid solutions. J. Phys. Chem. B. 1997;101:5405. Google Scholar
- Christmann K, Ertl G, Pignet T. Adsorption of hydrogen on a Pt(111) surface. Surface Science. 1976;54(2):365-392. Google Scholar
- Jerkiewicz G. Hydrogen sorption ATIN electrodes. Progress in Surface Science. 1998;57(2):137-186. Google Scholar
- Engstrom JR, Tsai W, Weinberg WH. The chemisorption of hydrogen on the (111) and (110)‐(1×2) surfaces of iridium and platinum. The Journal of Chemical Physics. 1987;87(5):3104-3119. Google Scholar
- Lasia A. Modeling of hydrogen upd isotherms. Journal of Electroanalytical Chemistry. 2004;562(1):23-31. Google Scholar
- Markovic NM, Ross PN. Surface science studies of model fuel cell electrocatalysts. Surface Science Reports. 2002;45(4-6):117-229. Google Scholar
- Hanh TTT, Takimoto Y, Sugino O. First-principles thermodynamic description of hydrogen electroadsorption on the Pt(111) surface. Surface Science. ;625:104-111. Google Scholar
- Kita H. Horiuti's generalized rate expression and hydrogen electrode reaction. Journal of Molecular Catalysis A: Chemical. 2003;199(1-2):161-174. Google Scholar
- Kunimatsu K, Senzaki T, Samjeské G, Tsushima M, Osawa M. ydrogen adsorption and hydrogen evolution reaction on a polycrystalline Pt electrode studied by surface-enhanced infrared absorption spectroscopy. Electrochimica Acta. 2007;52(18):5715-5724. Google Scholar
- Henry CR. Surface studies of supported model catalysts. Surface Science Reports. 1998;31(7-8):231-325. Google Scholar
- Dahl S, Logadottir A, Egeberg RC, Larsen JH, Chorkendorff I, Törnqvist E, et al. Role of steps in N2 activation on Ru(0001). Physical Review Letters. 1999;83(9):1814-1817. Google Scholar
- Hvolbaek B, Janssens TVW, Clausen BS, Falsig H, Christensen CH, Norskov JK. Catalytic activity of Au nanoparticles. Nano Today. 2007;2(4):14-18. Google Scholar
- Gudmundsdóttir S, Tang W, Henkelman G, Jónsson H, Skúlason E. Local density of states analysis using Bader decomposition for N2 and CO2 adsorbed on Pt(110)-(1×2) electrodes. The Journal of Chemical Physics. 2012;137(16):164705. PubMed Google Scholar
- Skúlason E, Karlberg GS, Rossmeisl J, Bligaard T, Greeley J, Jónsson H, Density functional theory calculations for the hydrogen evolution reaction in an electrochemical double layer on the Pt(111) electrode. Physical Chemistry Chemical Physics. 2007;9(25):3241-3250. PubMed Google Scholar
- Anger G, Berger HF, Luger M, Feistritzer S, Winkler A, Rendulic KD. Microfacets of the (1 × 2) reconstructed Pt(110) surface seen in the adsorption dynamics of H2. Surface Science Letters. 1989;219(3):L583-L589. Google Scholar
- Shern CS. Hydrogen adsorption on a Pt(110)−(1 × 2) surface. Surface Science. 1992;264(1-2):171-176. Google Scholar
- Minca M, Penner S, Dona E, Menzel A, Bertel E, Brouet V, et al. Surface resonances on transition metals as low-dimensional model systems. New Journal of Physics. 2007;9(10):386-386. Google Scholar
- Zhang Z, Minca M, Deisl C, Loerting T, Menzel A, Bertel E. H on Pt(110): An atypical chemisorption site at low coverages. Physical Review B. 2004;70(12):121401. Google Scholar
- Soler JM, Artacho E, Gale JD, García A, Junquera J, Ordejón P, et al. The SIESTA method forab initioorder-Nmaterials simulation. Journal of Physics: Condensed Matter. 2002;14(11):2745-2779. Google Scholar
- Ordejón P, Artacho E, Soler JM. Self-consistent order-Ndensity-functional calculations for very large systems. Physical Review B. 1996;53(16):R10441-R10444. PubMed Google Scholar
- Perdew J.P., Burke K., Ernzerhof M.. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996;77:3865. PubMed Google Scholar
- Gudmundsdóttir S, Skúlason E, Weststrate KJ, Juurlink L, Jónsson H. Hydrogen adsorption and desorption at the Pt(110)-(1×2) surface: experimental and theoretical study. Physical Chemistry Chemical Physics. 2013;15(17):6323. PubMed Google Scholar
- Waseda Y, Hirata K, Ohtani M. High-temperature thermal expansion of platinum, tantalum, molybdenum, and tungsten measured by x-ray diffraction. High Temp. - High Pressures. 1975;7(2):221-226. Google Scholar





